
Por David Orenstein | Instituto Picower para el Aprendizaje y la Memoria
Si bien muchos estudios abordan el síndrome de Rett, un trastorno del desarrollo, como una afección única derivada de la pérdida general de función del gen MECP2, un nuevo estudio realizado por neurocientíficos del Instituto Picower para el Aprendizaje y la Memoria del MIT demuestra que dos mutaciones diferentes de este gen causaron numerosas anomalías distintas en cultivos celulares de laboratorio. Además, corregir las diferencias clave producidas por cada mutación requirió tratamientos diferentes.
Mriganka Sur, autor principal del nuevo estudio de acceso abierto publicado en Nature Communications y profesor Newton en el Instituto Picower y el Departamento de Ciencias Cerebrales y Cognitivas. El estudio empleó cultivos avanzados de tejido cerebral humano en 3D, denominados «organoides» o «minicerebros», derivados de células de la piel o de la sangre donadas por pacientes con síndrome de Rett portadores de cada mutación. El autor principal, Tatsuya Osaki, científico investigador del Instituto Picower, afirma que la capacidad de los organoides para modelar las consecuencias específicas de cada mutación le permitió obtener información específica sobre cada mutación que no se había detectado en estudios anteriores, donde los científicos simplemente inactivaban el gen MECP2 en general. Los organoides también brindaron una oportunidad novedosa para comprender cómo cada mutación afectaba a los diferentes tipos de células y sus interacciones.
Efectos distintos
Más de 800 mutaciones en el gen MECP2 pueden causar el síndrome de Rett, pero solo ocho representan más del 60 % de los casos. Sur y Osaki eligieron una de ellas, la R306C, que implica una diferencia de tan solo un par de bases de ADN (916C>T), porque representa entre el 7 % y el 8 % de los casos de síndrome de Rett. La otra mutación que eligieron, la V247X, es mucho más rara y grave porque interrumpe la producción de la proteína del gen mediante la deleción de una sola base de ADN (705Gdel), lo que provoca que la proteína no solo sea defectuosa, sino incompleta.
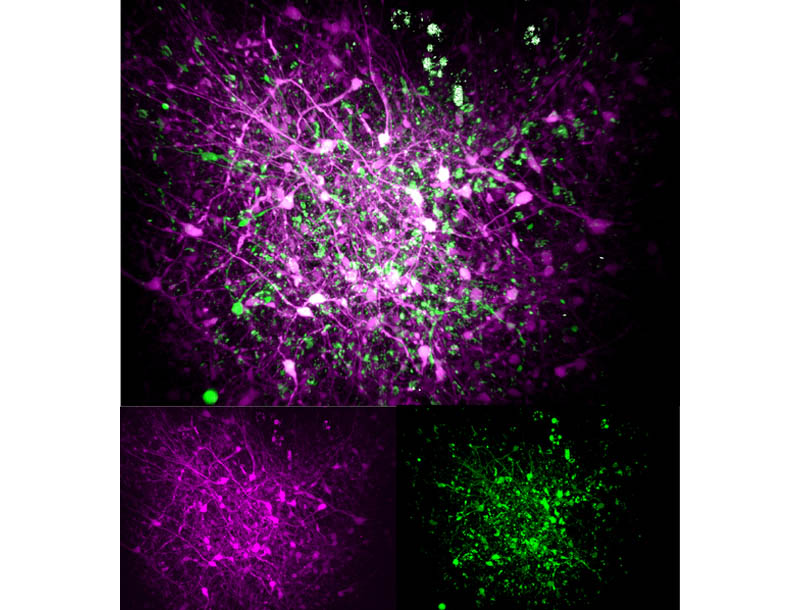
En organoides cultivados durante tres meses, cada mutación produjo algunas consecuencias comunes, pero también, en ocasiones, distintas, en comparación con los organoides de control con MECP2 no mutado. Para muchos de sus experimentos, el equipo utilizó microscopios de «tres fotones» capaces de alcanzar una resolución a nivel celular a lo largo de los organoides, cuyo grosor aproximado es de 1 milímetro, lo que permitió visualizar tanto su estructura (mediante imágenes de «generación de tercer armónico») como los patrones de actividad en vivo de sus neuronas (mediante fluorescencia de calcio).
Por ejemplo, los científicos observaron que los organoides V247X presentaban varias diferencias estructurales con respecto a sus controles: eran más grandes y tenían distintos grosores en las diferentes capas. En cambio, los organoides R306C se parecían mucho más a sus controles. Los organoides que presentaban cualquiera de las dos mutaciones mostraban proyecciones axónicas menos desarrolladas en sus neuronas, en comparación con sus homólogos de control.
Al analizar las propiedades de la actividad neuronal y la conectividad en los organoides, los científicos encontraron déficits similares en ambas mutaciones. Ambas mostraron una actividad de potenciales de acción y una sincronicidad entre neuronas reducidas en comparación con sus controles.
Pero al analizar otras propiedades, los organoides comenzaron a divergir entre sí. En particular, un indicador de la eficiencia de su estructura de red, denominado «propensión al mundo pequeño» (SWP, por sus siglas en inglés), disminuyó en los organoides R306C y aumentó en los V247X, en comparación con los controles. Esto significa que ambas mutaciones alteraron el desarrollo de las estructuras de red típicas para el procesamiento de información, pero en direcciones opuestas.
Para garantizar que sus resultados fueran relevantes para los pacientes con síndrome de Rett, el equipo colaboró con Charles Nelson del Boston Children’s Hospital, cuyo equipo midió el EEG en varios niños con diferentes mutaciones de Rett. Aunque la muestra era pequeña, los investigadores observaron indicios de que la propiedad SWP en las lecturas del EEG estaba alterada en los voluntarios, de forma similar a como ocurría en los organoides.
Finalmente, al marcar las neuronas excitatorias para que parpadearan en un color y las neuronas inhibitorias para que parpadearan en un color diferente, los científicos pudieron observar que la conectividad entre los diferentes tipos de neuronas difería significativamente de los controles en los organoides V247X.
Pruebas de tratamiento
Todas las pruebas demostraron que cada mutación provocaba varios cambios en la estructura, la actividad y la conectividad de los organoides, y que las desviaciones solían ser particulares de la mutación específica.
Para comprender cómo surgieron estas diferencias y cómo podrían corregirse, el equipo de Sur y Osaki examinó cómo las células de cada tipo de organoide expresaban sus genes de manera diferente a los controles. Las diferencias en la expresión génica suelen provocar alteraciones en vías moleculares clave de las células, lo que puede interrumpir su actividad y función. El análisis mediante una técnica denominada secuenciación de ARN de célula única reveló cientos de diferencias en cada tipo de organoide, donde algunos genes se expresaban más que en los controles, mientras que otros se expresaban menos.
Por ejemplo, los análisis revelaron que en los organoides R306C se sobreexpresaba un gen llamado HDAC2. Se sabe que esta proteína reprime la expresión de otros genes. Mientras tanto, en los organoides V247X, los científicos encontraron una expresión reducida de genes para algunos receptores del neurotransmisor inhibidor GABA. Estos organoides también mostraron defectos en la función de las células astrocíticas, que sustentan muchos aspectos de la función neuronal.
Los organoides con cualquiera de las dos mutaciones también mostraron aberraciones en las vías moleculares que permiten el desarrollo de conexiones de circuitos entre neuronas, llamadas sinapsis.
Dados los defectos específicos que observaron, los científicos decidieron tratar los organoides con un fármaco que inhibe la actividad de la HDAC2 y otro que aumenta la eficacia del GABA. El inhibidor de la HDAC2 restauró la actividad neuronal y el SWP a niveles normales en los organoides R306C, y el agonista del GABA, baclofeno, restauró el SWP a niveles de control en los organoides V247X.
Osaki señala que cada uno de los fármacos utilizados en el tratamiento ya ha sido estudiado en otros contextos de enfermedades, lo que significa que son medicamentos bien conocidos que podrían reutilizarse.
Ahora que los investigadores han desarrollado una plataforma de organoides para analizar las consecuencias de mutaciones individuales, identificando tanto sus orígenes como probando tratamientos, planean aplicarla al estudio de cuatro mutaciones más, explica Sur, comparándolas todas con un organoide de control estandarizado.
Además de Sur, Osaki y Nelson, los otros autores del artículo son Chloe Delepine, Yuma Osako, Devorah Kranz, April Levin y Michela Fagiolini.
